paired end sequencing insert size
Sequence information from two ends of a short DNA fragment usually a few hundred base pairs long. Instead of building a bimodal distribution of plus and minus strand reads to predict fragment size MACS2 will use actual insert sizes of pairs of reads to build fragment pileup.

Stacks 2 Analytical Methods For Paired End Sequencing Improve Radseq Based Population Genomics Biorxiv
These are approximations only as the actual fragment size distribution will depend on a number of factors including the type.
. For all paired-end sequencing we recommend using HMW Buffer only. Fragment length150bp two mates are in length of 100bp. What should I do if the two mates have overlaps.
Paired-end sequencing enables both ends of the DNA fragment to be sequenced. COVIDSeq Assay 96 samples Illumina DNA Prep. 6 Paired-end sequencing alignment QA61.
Each kit is processed as a single batch and is not. Because the distance between each paired read is known. Dec 19 2012 ericminikel.
Such tools include SeqPrep Stitch etc. Any recommendation for mapping paired-end BS-seq data. The sequencing occurs for millions of clusters at once and each cluster has 1000 identical copies of a DNA insert.
2 read with mate that is aligned to a different chromosome. Libraries with the same rank would be used at the same time. Paste the raw or FASTA sequence into the text area below.
The distribution is somewhat leptokurtic and positively skewed with a. If your reads are paired the application additionally calculates insert size statistics such as median and mean insert sizes median absolute deviation and standard deviation of. They separate and the 3 end of each strand is blocked.
The current script is designed to work with Paired End data Due to HPC queue limits on mercer the pipeline can run on a maximum of 25 libraries at a time If you need to run the pipeline on more than 25 libraries or on Single End reads. IGV colors 1 paired end reads with inferred insert size smaller or larger than expected. 3 paired-end alignments with deviant pair orientation.
Although both platforms can generate single-end and paired-end sequencing reads the maximum read length is just 75 bp for SOLiD and 28100 bp for Complete Genomics 33 limiting their use for. Illumina relies on paired-end reads to extend outside the bait sequences and fill in the gaps Clark MJ. Forward and reverse reads in paired-end sequencing.
I suggest a pre-step for merging two overlapped reads into one. Match your chosen kit size to the number of samples you run at a time. For BWA at least the proper pair flag depends not only on the FR orientation but also on insert size being within a certain range from BWA manual.
Left and right position of the fragment from Paired-end sequencing. Fast high-quality sample-to-data next-generation sequencing services. The distribution shows a peak insert size of around 300 bp.
To obtain paired reads separated by larger distances we circularized DNA fragments of the required length for example 2 02 kb and obtained short junction fragments for paired end sequencing. We run 2 100 paired-end reads and our exome sequencing libraries typically contain insert sizes of approximately 250 bases in length as a compromise to match the average size of most exons while sequencing without overlapping read pairs. Poplar was chosen as the first tree DNA to be sequenced because of its relatively compact genetic complement some 50 times smaller than the genome of pine making the poplar an ideal model system for trees.
Fragments can consist of single reads typically 501000 bp or of paired-end reads of varying insert size note that paired-end reads can even overlap. This website uses cookies to help provide you with the best possible online experience. The maximum distance x for a pair considered to be properly paired SAM flag 02 is calculated by.
Many sequencing library preparation kits include an option to generate so-called. The size of sequencing tags. If you dont specify it MACS will try to use the first 10.
For example in a dataset of a human genome we set five ranks for five libraries with insert size 200-bp 500-bp 2-Kb 5-Kb and 10-Kb separately. Please read our Terms Conditions and Privacy Policy for information about. The forward strand is washed away and the process of sequence by synthesis repeats for the reverse strand.
Note that coloring by insert size is a feature designed originally for DNA alignments against the genome. The size of an RNA-Seq library is also determined by the applications. SOAPdenovo will use the pair-end libraries with insert size from smaller to larger to construct scaffolds.
Whole-exome sequencing data analysis. During library preparation sample DNA is fragmented and the fragments of a specific size typically 200500 bp but can be larger are ligated or inserted in between two oligo adapters. It is based on set base pair.
There is a unique adapter sequence on both ends of the paired-end read labeled Read 1 Adapter and Read 2 Adapter. Populus trichocarpa was sequenced ten times over to attain the highest quality standards and to produce a relatively contiguous high quality plant genome. Mate-pair libraries span larger genomic regions 220 kb.
Fragment Size approx Insert Size approx LMW Buffer HMW Buffer LMW Buffer HMW Buffer Lambda DNA Control 175-400 bp 175-700 bp 40-265.
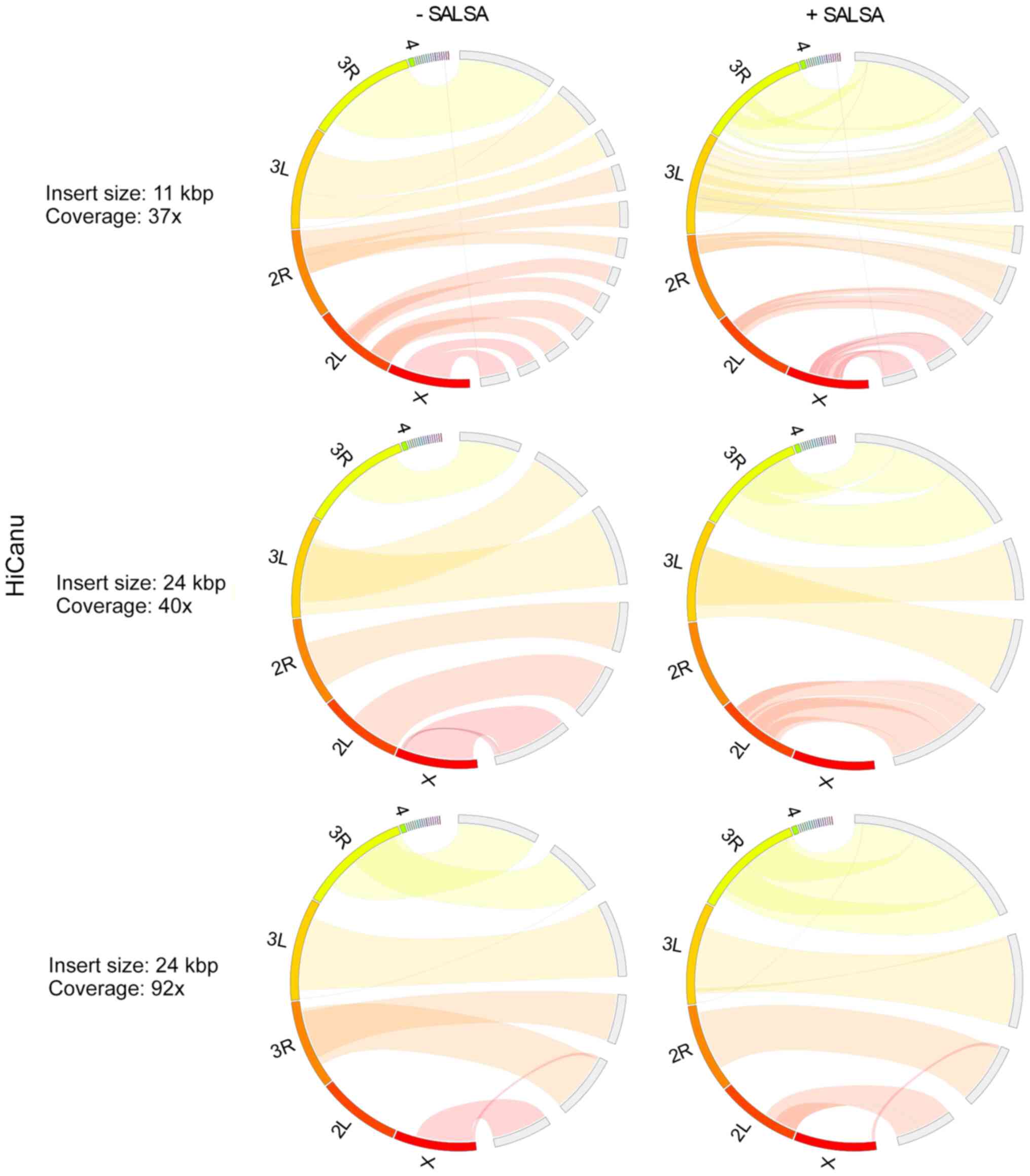
Benchmarking Of Next And Third Generation Sequencing Technologies And Their Associated Algorithms For Em De Novo Em Genome Assembly
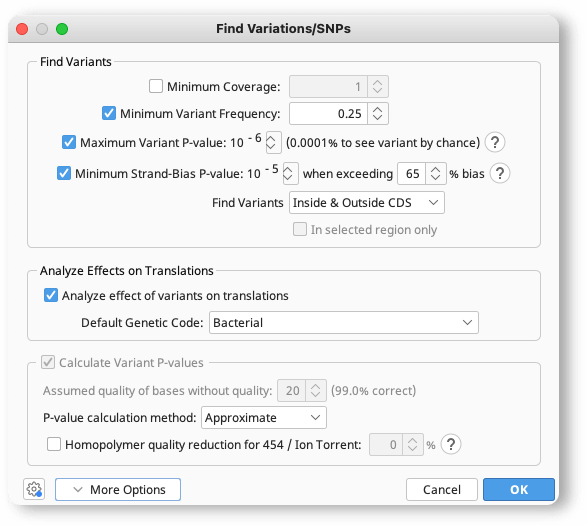
Mapping And Snp Calling Tutorial Geneious Prime

Sequencing By Ligation An Overview Sciencedirect Topics

Next Generation Sequencing Tips N Tricks Part 4 Diagnostech
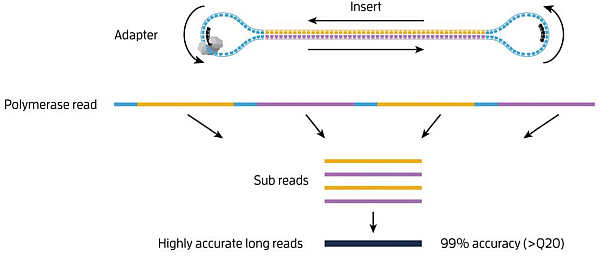
Ucd Bioinformatics Core Workshop

Stacks 2 Analytical Methods For Paired End Sequencing Improve Radseq Based Population Genomics Biorxiv
2

Mapping And Snp Calling Tutorial Geneious Prime
A Robust Simple Genotyping By Sequencing Gbs Approach For High Diversity Species Plos One
2
2

Stacks 2 Analytical Methods For Paired End Sequencing Improve Radseq Based Population Genomics Biorxiv

Stacks 2 Analytical Methods For Paired End Sequencing Improve Radseq Based Population Genomics Biorxiv

Improved Protocols For Illumina Sequencing Bronner 2013 Current Protocols In Human Genetics Wiley Online Library
More Information How To Calculate Meanfragmentlength Issue 1 Aalhendi1707 Counttofpkm Github
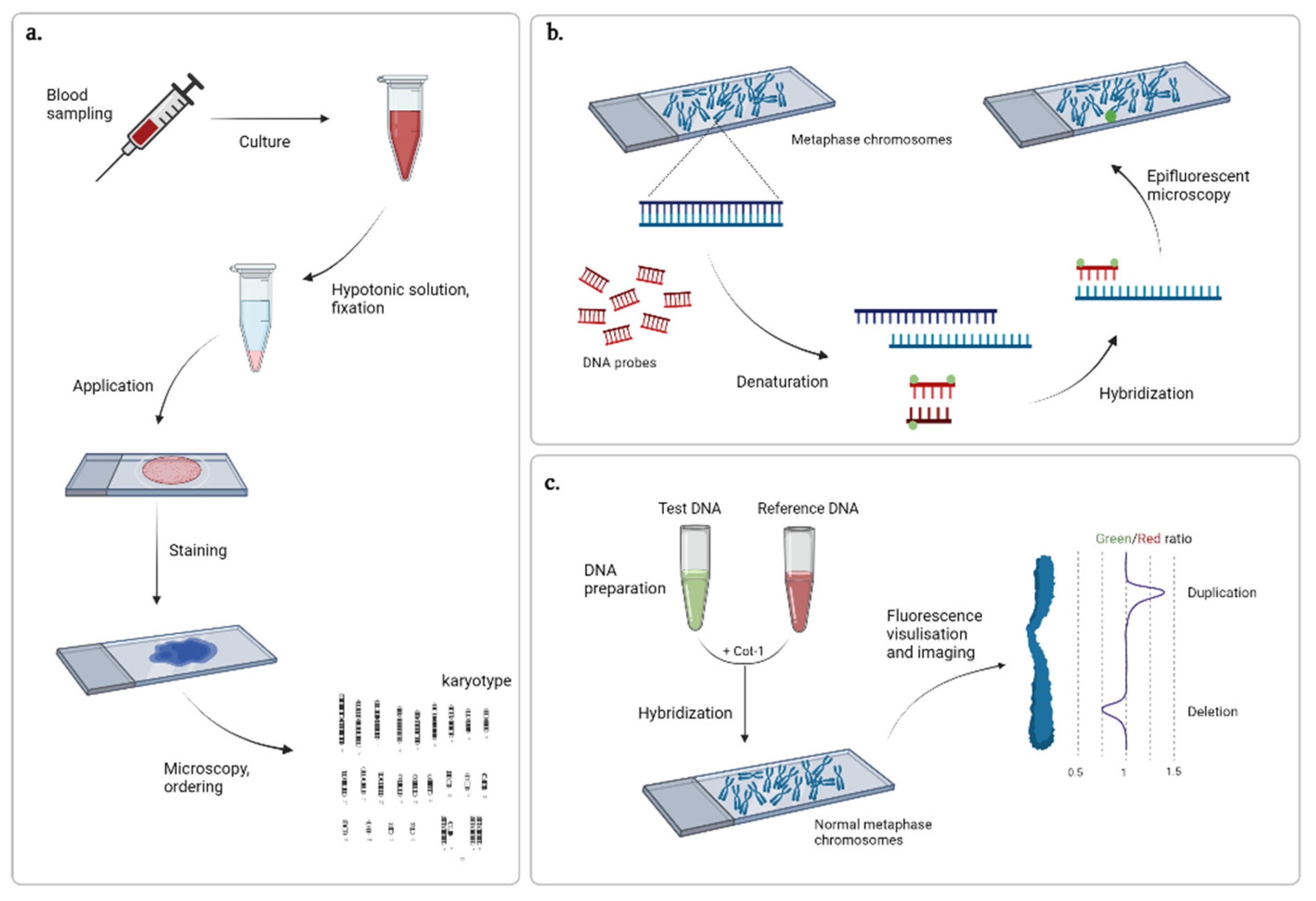
Ijms Free Full Text Progress In Methods For Copy Number Variation Profiling Html

Mapping And Snp Calling Tutorial Geneious Prime

Gbs Adapters Pcr And Sequencing Primers A Sequences Of Download Scientific Diagram

Improved Protocols For Illumina Sequencing Bronner 2013 Current Protocols In Human Genetics Wiley Online Library